FDA news and Views: Quality Overall Summary, Third Party Review Metrics, Rogue Online Pharmacies, State of CDER, Warning for illegal, unapproved opioid cessation products
FDA BRIEF: Week of January 22, 2018

Quality Overall Summary (QOS) of all quality-related information provided in NDA, ANDA, BLA
- Considering adjustments to QOS format to improve efficiency
CDER new white paper describes key considerations for QOS preparation:
- Explaining product and process development in a patient-focused context
- Effectively summarizing the overall control strategy
- Guiding the regulator through the submission
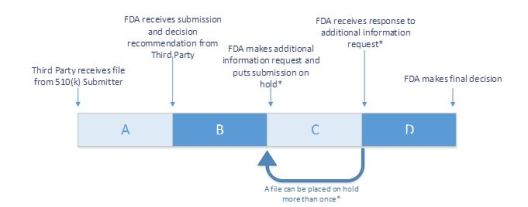
510(k) Third Party Review Metrics
510 (k) Accredited Persons Program created to improve efficiency and timeliness
- FDA accredits Third Parties to conduct 510(k) primary review
- Third Party 510(k) submission goes through four different stages
- Stage A – Reviews sponsor submission and sends recommendation to FDA
- Stage B – FDA reviews submission to ensure Third Party has submitted
all information needed to make final decision - Stage C (Optional) – Third Party reviews FDA’s request for additional
information and notifies 510(k) submitter - Stage D – FDA reviews additional information and makes final decision.
Accredited Organizations: AABB, CMSI: Center for Measurement Standards of Industria, NYSDOH: New York State Department of Health, NIOM: Nordic Institute of Dental Materials, RTS: Regulatory Technology Services, LLC, TPRG: Third Party Review Group, LLC, TUV: TUV SUD America Inc


Buying from Online Pharmacies
Rogue online pharmacies
- No valid prescription required
- Do not have U.S. state-licensed pharmacist to answer questions
- Low prices seem too good to be true
- Send spam or unsolicited email offering cheap medicine
- Located outside of US or ship worldwide.
Sell dangerous products with compromised safety and effectiveness
- Under dosage or over dosage
- Incorrect active ingredient
- Addition of unsafe ingredients
- Incorrect storage
Shop Safely Online
- Learn
- Report rogue pharmacies to FDA
- Report adverse effects to FDA’s MedWatch
- FDA’s web page on counterfeit medicine for more information
 Podcast: State of CDER
Podcast: State of CDER
Podcast: State of CDER 2018, Transcript

FDA, FTC warn companies for selling illegal, unapproved opioid cessation products using deceptive claims
FDA + FTC joint warning letters to marketers and distributors of 12 opioid cessation products
- Health fraud for illegally marketing unapproved products with claims about treatment of opioid addiction and withdrawal
- Products have not been demonstrated to be safe or effective
- May keep patients from seeking appropriate, FDA-approved therapies
- Making unsubstantiated therapeutic claims is violation of the Federal Trade Commission Act
Have requested responses from each of the companies within 15 working days
- Specific actions taken to address each concern
- Failure to correct violations may result in law enforcement action such as seizure or injunction
Image credit: FDA
|
|
|||||||
|
|
|||||||
|
|||||||