Month: April 2018
2018 FDA CALENDAR
2018 FDA CALENDAR

⊕ New
AUGUST
Aug 13-14: Public Meeting – Pediatric Medical Device Development. Identify strategies that enhance medical device ecosystem toward development and innovation of devices that serve the complex needs of children. INFORMATION
JULY
⊕ Jul 9: Public Meeting for Patient Focused Drug Development for Chronic Pain. Hearing patients’ perspectives on chronic pain, treatment approaches, challenges or barriers to accessing treatments and managed with opioids, acetaminophen, NSAIDs, antidepressants, non-pharmacologic interventions or therapies.INFORMATION
JUNE
Jun 19-20: FDA/IMSN Summit with international drug regulators to discuss medication safety issues. Discussion among international drug regulators on medication safety issues with regulated pharmaceutical products and how to minimize medication errors with labeling and packaging. INFORMATION
Jun 22: Clinical Outcome Assessments (COAs) in Cancer Clinical Trials. Discussion between academia, industry, international regulatory, HTA bodies, and patient groups to advance measurement of the patient experience in cancer clinical trials; characteristics of PRO measurement tools, standardize PRO data analysis, FDA approaches to PRO data review. INFORMATION
Jun 25-26: 2018 Center for Biologics Evaluation and Research Science Symposium. Discuss scientific topics related to the regulation of biologics and highlight science conducted at CBER by showcasing how scientific research informs regulatory decision making and to provide a forum for developing collaborations within FDA and with external organizations.
MAY
⊕ May 2: Antimicrobial Drugs Advisory Committee Meeting. Discuss NDA for plazomicin, sponsored by Achaogen for proposed indications for treatment of complicated urinary tract infections & blood stream infections in adults INFORMATION
⊕ May 3: Joint Meeting of the Gastrointestinal Drugs Advisory Committee and the Pediatric Advisory Committee Meeting. Discuss NDA for stannsoporfin injection, submitted by InfaCare, proposed for treatment of neonates with indicators of hemolysis who are at risk of developing severe hyperbilirubinemia INFORMATION
⊕ May 4: Annual Public Meeting; Reagan-Udall Foundation for the FDA. FDA Commissioner Scott Gottlieb as keynote speaker; discuss activities and how it supports FDA; panel discussion on “Evolution of FDA Science and Engagement.” INFORMATION
⊕ May 8: FDA Drug Topics: Protecting Patients – Pharmacists Requirements under the Drug Supply Chain Security Act. FDA’s Division of Drug Information in the Center for Drug Evaluation and Research (CDER) is presenting a series of continuing education webinars targeting the needs of all health care professionals. INFORMATION
May 8: CBER Update: Office of Vaccine Research and Review Data Submission. Update on best practices INFORMATION
⊕ May 9: Tissue Agnostic Therapies: Regulatory Considerations for Orphan Drug Designation; Public Workshop. Discuss factors when evaluating drugs for orphan designation that treat tissue agnostic disease or condition in oncology, additional factors related to orphan exclusivity when approving such a product INFORMATION
⊕ May 10: User Session – Digital Health Software Precertification (Pre-Cert) Pilot Program. Interactive session to discuss progress on Software Precertification Pilot Program, working model, key program areas, questions for public input. INFORMATION
⊕ May 10: FDA Grand Rounds spotlights stakeholder opportunities in FDA predictive toxicology roadmap. Framework for integrating novel predictive toxicology methods into safety and risk assessments of its products. INFORMATION
May 10: Endocrinologic and Metabolic Drugs Advisory Committee Meeting. Discuss NDA for volanesoren solution for subcutaneous injection, Akcea Therapeutics, Inc, for use as an adjunct to diet for the treatment of patients with familial chylomicronemia syndrome. INFORMATION
May 11: Medical Gas Workshop III. Provide opportunity for medical gas manufacturers and public to provide input on potential areas of Federal drug regulation that should be revised with respect to medical gases. INFORMATION
May 15-16: FDA Regulatory Education for Industry (REdI). FDA-led forum that brings together the regulatory educators from FDA’s CDER and CDRH. Meeting Information
⊕ May 17: Webinar: Pioneering Modeling Methodologies in Generic Drug Development. Discuss novel ways FDA is approaching the use of quantitative methods and modeling for development and demonstration of generic sameness. INFORMATION
May 18: MDIC workshop on Patient-Centered Clinical Trial Design. Method for incorporating patient preference information to set significance levels in clinical trial design. Focus on Parkinson’s disease; may be generalizable to other diseases. In collaboratorion with FDA (CDRH), MIT, RTI Health Solutions and Michael J. Fox Foundation. INFORMATION
⊕ May 22: Joint Meeting of the Anesthetic and Analgesic Drug Products Advisory Committee and the Drug Safety and Risk Management Advisory Committee. Discuss NDA for buprenorphine sublingual spray, by INSYS, for the treatment of moderate-to-severe acute pain where the use of an opioid analgesic is appropriate INFORMATION
May 22-23: Accreditation Scheme for Conformity Assessment (ASCA) of Medical Devices to Food and Drug Administration-Recognized Standards; Public Workshop. Discuss draft design of pilot program, including goals and scope, framework, requirements, and streamline standards. INFORMATION
May 24: FY 2018 Generic Drug Research Public Workshop. Provide overview of regulatory science initiatives for generic drugs and public input on research priorities INFORMATION
APRIL
Apr 3: Public workshop: CDER and You: Keys to Effective Engagement. Share information with stakeholders including patients, patient advocates, academic and medical researchers, health care professionals, drug developers. Share unique perspectives on drug development and safety. INFORMATION
Apr 6: US FDA and Health Canada: Joint Regional Consultation on the ICH. To provide information and receive comments on the current ICH activities as well as the upcoming meetings in Kobe, Japan INFORMATION
Apr 10: FDA Drug Topics: An Introduction to Drug Safety Surveillance and the FDA Adverse Event Reporting System. Introduce the many phases of drug safety surveillance from the earliest stages of drug development through post approval, and will focus on how FDA conducts pharmacovigilance, develops safety signals, and communicates our findings.INFORMATION
Apr 11-12: CDER Small Business and Industry Assistance Regulatory Education for Industry (REdI): Generic Drugs Forum. Interact with FDA subject matter experts on Generic Drug Review Program, program progress, current initiatives. INFORMATION
Apr 16: Evaluating Inclusion and Exclusion Criteria in Clinical Trials; Public Meeting. Discuss topics related to eligibility criteria in clinical trials and their potential impact on patient access to investigational drugs, and how to facilitate the enrollment of a diverse patient population. INFORMATION
Apr 16: Public Workshop: Study Design Considerations for Devices including Digital Health Technologies for Sleep Disordered Breathing (SDB). Appropriate design of clinical studies to evaluate devices including digital health technologies intended for the diagnosis, monitoring, or treatment of SDB INFORMATION
Apr 16-18: 2018 AAPS Workshop on Drug Transporters in ADME: From Bench to Bedside. Present next generation of transporters and transport mechanisms that may contribute to ADME properties of drugs in disease states considered in drug discovery and development. INFORMATION
Apr 17: Public Meeting on Patient-Focused Drug Development for Opioid Use Disorder (OUD). To learn patients’ perspectives on OUD, effects on health and well-being, experience using prescription medical treatments and other treatments, challenges or barriers to accessing medical treatments for OUD. INFORMATION
Apr 19: Peripheral and Central Nervous System Drugs Advisory Committee Meeting Discuss NDA for cannabidiol oral solution, GW Pharmaceuticals, for the adjunctive treatment of seizures associated with Lennox-Gastaut syndrome or Dravet syndrome in patients 2 years of age and older. INFORMATION
Apr 20: FDA/OCE Public Meeting on Relevant Molecular Targets in Pediatric Cancers: Applicability to Therapeutic Investigation FDARA 2017. Discuss provisional list of molecular targets for adult cancer indications but also relevant to cancer in children thereby providing a rationale for early pediatric evaluation. INFORMATION
Apr 21: Public Meeting on Electronic Submissions and Data Standards. Discuss current status of electronic submissions and data standards initiatives to improve predictability and consistency of electronic submissions process in support of human drug review program. INFORMATION
Apr 23: Arthritis Advisory Committee Meeting. Discuss NDA for baricitinib tablets, submitted by Eli Lilly and Company, for the treatment of adult patients with moderately to severely active rheumatoid arthritis who have had an inadequate response or intolerance to methotrexate. INFORMATION
Apr 23-25: 12th Annual FDA/DIA Biostatistics Industry and Regulator Forum. Discuss relevant statistical issues associated with the development and review of therapeutic drugs and biologics. INFORMATION
Apr 30. Public Workshop – Orthopaedic Sensing, Measuring, and Advanced Reporting Technology (SMART) Devices. Enhance engagement with stakeholders to facilitate device development and to discuss scientific and regulatory challenges associated with Orthopaedic SMART Devices. INFORMATION
MARCH
Mar 1: Vaccines and Related Biological Products Advisory Committee Meeting. Hear research program in the Laboratory of Mucosal Pathogens and Cellular Immunology (LMPCI), Division of Bacterial, Parasitic and Allergenic Products (DBPAP), Office of Vaccines Research and Review (OVRR), and discuss selection of strains to for vaccines for the 2018-2019 influenza season INFORMATION
Mar 1: 21st US-Japan Cellular and Gene Therapy Conference. Exchange ideas on cutting edge and diverse areas of biomedical research, and enhance opportunities for collaborations among scientists from the US and Japan. INFORMATION
Mar 1: Neurological Devices Panel Advisory Committee Meeting. Discuss safety and effectiveness of intracranial aneurysm treatment devices and factors affecting clinical outcomes such as aneurysm morphology, size, and location in the neurovasculature. INFORMATION
Mar 1-2: IASLC-FDA Lung Cancer Neoadjuvant Meeting.Discuss standardization and validation of endpoints in neoadjuvant lung cancer trials. INFORMATION
Mar 4-6: FDA-PhUSE Computational Science Symposium. Review progress on topics such as data standards, best-practices-driven analytical tool development, business processes for information systems, evaluation of current tools. INFORMATION
Mar 5: Risk Communication Advisory Committee Meeting. Committee will discuss impact of pregnancy and lactation labeling information in prescription drug and biological products as modified under the Pregnancy and Lactation Labeling Rule. INFORMATION
Mar 8: FDA Grand Rounds: Stem cell-based cellular therapies. Use of stem cell-based products is new and characterizing the product still faces hurdles. FDA conducting research into identifying cell therapy product characteristics that will predict the reliably of the performance of cell-based therapies in humans. INFORMATION
Mar 8: Public Workshop: Safety Assessment for Investigational New Drug Safety Reporting. Engage external stakeholders in discussions related to finalizing the draft guidance entitled “Safety Assessment for IND Safety Reporting.” INFORMATION
Mar 15: Oncology Center of Excellence Listening Session; Solicit comments regarding what stakeholders desire of the OCE in terms of structure, function, regulatory purview, and activity. INFORMATION
Mar 20: Promoting the Use of Complex Innovative Designs (CID) in Clinical Trials. March 20, 2018. Discuss use of CID in drug development and regulatory decision making, CID pilot program. INFORMATION
Mar 21-22: Joint Meeting of the Blood Products Advisory Committee and the Microbiology Devices Panel. Discuss reclassification from Class III to Class II of nucleic acid and serology-based point-of-care and laboratory-based in vitro diagnostic devices indicated for use as aids in the diagnosis of human immunodeficiency virus (HIV) and hepatitis C virus (HCV) infection.⊕ INFORMATION
Mar 22: Webinar – Duodenoscope Sampling and Culturing. FDA, CDC, ASM and other endoscope culturing experts will review the voluntary duodenoscope surveillance sampling and culturing protocols to monitor quality of reprocessing procedures. INFORMATION
Mar 22: Patient Engagement in the National Evaluation System for health Technology (NEST): Lessons Learned and Best Practices Workshop. Gather lessons learned and best practices for patient engagement in evidence generation (planning, collection of data and information, analysis, and dissemination). INFORMATION
Mar 22: Pediatric Advisory Committee and Endocrinologic and Metabolic Drugs Advisory Committee Meeting. To discuss major objectives of Phase 3 drug development program for treatment of children with achondroplasia (ACH) submitted by BioMarin Pharmaceutical Inc. INFORMATION
Mar 23: Advisory Committee Meeting: Pediatric. Discuss the following products for CDER – BANZEL, INTUNIV, LEXAPRO and CDRH – FLOURISH, ACTIVA, LIPOSORBER, IMPELLA RP SYSTEM. INFORMATION
Mar 26-28: Gastroenterology Regulatory Endpoints and the Advancement of Therapeutics for Alcoholic Hepatitis and Alcohol Associated Liver Disease and Pediatric Irritable Bowel Syndrome and Pediatric Functional Constipation Workshop, Facilitate dialogue among industry, academia, and other stakeholders on common data elements needed to be included in clinical trials, clinical trial designs, potential surrogate and clinical benefit endpoints, and practical issues with managing clinical trials. INFORMATION
Mar 27: Meeting of the Psychopharmacologic Drugs Advisory Committee. Discuss NDA for lofexidine hydrochloride, US WorldMeds, LLC, for mitigation of symptoms associated with opioid withdrawal and facilitation of completion of opioid discontinuation treatment. INFORMATION
Mar 28: Promoting the Use of Complex Innovative Designs in Clinical Trials
Inform development of guidance document and CID pilot program. INFORMATION
FEBRUARY
Feb 1: FDA-ISoP Public Workshop: Model Informed Drug Development (MIDD) for Oncology Products. Discuss integrating human pharmacokinetic, pharmacodynamic, efficacy, safety data into models, use of novel imaging techniques, diagnostic, predictive biomarkers, shift from traditional endpoints, regulatory implications. INFORMATION
Feb 7-8: 10th Annual Sentinel Initiative Public Workshop. Bring stakeholder community together to discuss a variety of topics on active medical product surveillance. INFORMATION
Feb 14-15: Joint Drugs Advisory Committee Meeting: Anesthetic and Analgesic Products and Drug Safety and Risk Management. Discuss NDA for HYDEXOR, for the short-term management of acute pain severe enough to require an opioid analgesic while preventing and reducing opioid-induced nausea and vomiting. Also discuss sNDA for EXPAREL (bupivacaine liposomal injectable suspension) to produce local analgesia and as a nerve block to produce regional analgesia. INFORMATION
Feb 22-23: FDA-AACR-ASTRO Regulatory Science and Policy Workshop – Clinical Development of Drug-radiotherapy Combinations. Address the lack of drug development for products intended specifically for use with radiation therapy. INFORMATION
Feb 27: Webinar – Custom Device Annual Reporting. Custom Device Exemption allows manufacturers to market medical devices designed to treat a unique pathology or physiological condition without premarket approval. Webinar to discuss custom device annual report requirement. INFORMATION
Feb 28: Public Meeting: Enhanced Drug Distribution Security under the Drug Supply Chain Security Act.Provide members of the drug distribution supply chain and other interested stakeholders an opportunity to discuss strategies and issues related to the enhanced drug distribution security provisions of the Act. INFORMATION
JANUARY
Jan 8: CLIA Waiver Applications Draft Guidance Documents. Discuss draft guidances on CLIA waiver applications and Dual 510(k) and CLIA waivers INFORMATION
Jan 9: Webinar – Pediatric Information for X-ray Imaging Device Premarket Notifications: Discuss final guidance on radiation safety of pediatric populations in the design of X-ray imaging devices. INFORMATION
Jan 10: Webinar – Technical Considerations for Additive Manufactured Medical Devices. Technical aspects associated with AM processes, recommendations for device design, manufacturing, testing considerations. INFORMATION
Jan 11: Public Workshop – Self-Collection Devices for Pap Test. Obtain feedback on feasibility, benefits, risks for self-collection cervical sampling devices for cervical cancer screening by Pap testing INFORMATION
Jan 11: Safety Assessment for IND Safety Reporting.Convened by the Duke-Robert J. Margolis Center for Health Policy at Duke University and FDA; to bring stakeholder community together to discuss IND safety topics INFORMATION
Jan 11: FDA Grand Rounds. Marker of brain injury increased in African Americans with Alzheimer’s. Better understanding of ethnicity and gender differences involved in the cause and progression of Alzheimer’s Disease could contribute to better drugs–and other types of interventions–to slow Alzheimer’s progression INFORMATION
Jan 16: Webinar – FDA Categorization of Investigational Device Exemption (IDE) Devices to Assist the Centers for Medicare and Medicaid Services (CMS) with Coverage Decisions. Discuss final guidance on FDA categorization of IDE devices that is used by CMS to determine whether an IDE device, and certain related services, may be covered by CMS. INFORMATION
Jan 26: Evaluating Nicotine Replacement Therapies. Public comments on FDA’s approach to evaluating the safety and efficacy of nicotine replacement therapy (NRT) products, including how they should be used and labeled. INFORMATION
Jan 29: Weighing the Evidence: Variant Classification and Interpretation in Precision Oncology. To discuss how genetic sequencing data is best implemented in patient management to advance innovative regulatory strategies to support development of safe and effective precision-based drugs and devices. INFORMATION
Jan 30: Opioid Policy Steering Committee–Prescribing Information Receive stakeholder input on how FDA REMS authority, might improve the safe use of opioid analgesics by curbing over-prescribing to decrease the occurrence of new addictions and limit misuse and abuse INFORMATION
Jan 30-31: Fostering Digital Health Innovation. Developing the Software Precertification Program. Discuss progress of pilot precertification program and seek input on ongoing development of the Software Precertification Program. INFORMATION
Image credit: FDA
Digital Health Software Precertification (Pre-Cert) Program

Digital Health Software Precertification (Pre-Cert) Program
GOALS
- Develop tailored and pragmatic regulatory oversight
- Trust organizations with demonstrated culture of quality and organizational excellence
- Leverage transparency of organization’s excellence and product performance across entire lifecycle
- Streamline premarket review to verify the continued safety, effectiveness, and performance
INTENTIONS
- Leverage information from all available sources to be more efficient and streamlined without compromising safety and effectiveness
- Enable modern and tailored approach to allow timely software iterations and changes
- Ensure high-quality by enabling companies to demonstrate their embedded culture of quality and organizational excellence
- Learn, adapt, adjust key elements based on program effectiveness
Image credit: FDA
FDA Market Authorization: CRYSVITA, TAVALISSE, MALDI Biotyper, CARDIOFORM Septal Occluder, GUARDIAN Connect System

CRYSVITA (burosumab-twza) injection
Ultragenyx Pharmaceutical, Inc.
INDICATION: Treatment of X-linked hypophosphatemia (XLH) in adult and pediatric
patients 1 year of age and older.
ADDRESSING UNMET NEED:
- XLH, a rare, inherited form of rickets, affects ~ 3,000 children and 12,000 adults in US
- XLH differs from other forms of rickets in that vitamin D therapy is not effective
- First drug approved to treat adults and children ages 1 year and older with XLH
MECHANISM OF ACTION: Binds to and inhibits the biological activity of FGF23 restoring renal phosphate reabsorption and increasing the serum concentration of 1,25 dihydroxy vitamin D.
EFFICACY:
- 65 pediatric patients and 134 adults with XLH in 4 studies, CRYSVITA vs, placebo,
- Achievement of normal phosphorus levels: Adults: 94% adults vs. 8%, Children: 94-100%
- Improved radiographic evaluation of rickets vs. natural history cohort: In both children and adults,
SAFETY:
- Most common adverse reactions: Back pain, headache, restless leg syndrome, decreased vitamin D, dizziness and constipation
- Most common adverse reactions in children: Headache, injection site reaction, vomiting, decreased vitamin D and pyrexia
REGULATORY PATHWAY: BLA
- Orphan designation, Breakthrough therapy designation
- Pediatric Priority Review Voucher granted
- Exempt from required pediatric assessments
- Postmarketing requirements: Post-approval surveillance program with safety objectives, lactation sub-study in lactating women , reanalyze banked immunogenicity serum samples
REIMBURSEMENT PATHWAY: For rare disease indication
- Covered with limited issue in Medicaid and Medicare
- However, increased scrutiny of rare disease therapies and evolution of
precision medicine; need to articulate long-term benefit of early
diagnosis and treatment to patient and the health care system

TAVALISSE (fostamatinib disodium hexahydrate) tablets
Rigel Pharmaceuticals, Inc.
INDICATION: Treatment of thrombocytopenia in adult patients with chronic immune
thrombocytopenia (ITP) who have had an insufficient response to a previous treatment
EFFICACY:
- Two identical, double-blind, placebo-controlled trials, n=150, patients with persistent or chronic ITP who had an insufficient response to previous treatment, TAVALISSE vs. placebo
- Endpoint: Stable platelet response (at least 50 x109/L on at least 4 of the 6 visits between Weeks 14 to 24)
- Study 1: 18% (n=9) vs, 0% (p=0.03)
- Study 2: 16% (n=8) vs. 4% (n=1), (p=0.26)
- Study 3: Stable response in 23% (n=10)
- Durable platelet responses seen
SAFETY:
- Most common adverse reactions: Diarrhea, hypertension, nausea, dizziness, alanine aminotransferase/aspartate aminotransferase (ALT/AST) increased, respiratory infection, rash, abdominal pain, fatigue, chest pain, and neutropenia
- Serious adverse drug reactions: Febrile neutropenia, diarrhea, pneumonia, and hypertensive crisis
REGULATORY PATHWAY: NDA
- Orphan desugnation
- Postmarketing commitmenets: Quality assessments

MALDI Biotyper CA System
Bruker Daltonik GmbH
INDICATION FOR USE: Mass spectrometer system using matrix-assisted laser
desorption/ionization – time of flight (MALDI-TOF) for the identification and differentiation of microorganisms cultured from human specimens.
The MALDI Biotyper CA System is a qualitative in vitro diagnostic device indicated for use in conjunction with other clinical and laboratory findings to aid in the diagnosis of bacterial and fungal infections – particularly Candida auris (C. auris)
ADDRESSING UNMET NEED:
- Emerging pathogen Candida auris (C. auris) can cause serious infections in hospitalized patients
- Can cause serious infections in hospitalized patients (e.g., bloodstream infections) and is frequently resistant to multiple antifungal drugs used to treat Candida infections.
GENERIC DEVICE TYPE: Clinical mass spectrometry microorganism identification and differentiation system
- Qualitative in vitro diagnostic device intended for the identification and differentiation of microorganisms from processed human specimens. The system acquires, processes, and analyzes spectra to generate data specific to microorganism(s). The device is indicated for use in conjunction with other clinical and laboratory findings to aid in the diagnosis of bacterial and fungal infection.
TESTING:
- Evaluated use of a standard protocol for adding C. auris to system database in conjunction with the performance data of 28 C. auris isolates (samples)
- C.auris isolates obtained from various culture collections, including the U.S. Centers for Disease Control and Prevention’s and FDA’s Antibiotic Resistance Isolate Bank.
- System can reliably identify C. auris 100% of the time
RISKS AND MITIGATION MEASURES:
- Incorrect identification or lack of identification of a pathogenic microorganism: Special Controls
- Failure to correctly interpret test results: Special Controls
- Failure to correctly operate the instrument: Special Controls
REGULATORY PATHWAY: De Novo request
- Add to cleared uses for identification of 333 species or species groups, covering 424 clinically relevant bacteria and yeast species
- Regulation Number: 21 CFR 866.3378
- Regulation Name: Clinical Mass Spectrometry Microorganism Identification and Differentiation System
- Regulatory Class: Class II
- Product Code: QBN

GORE® CARDIOFORM Septal Occluder
W. L. Gore and Associates, Inc.
SUPPLEMENTAL INDICATION FOR USE: Permanently implanted device indicated for the percutaneous, transcatheter closure of the following defects of the atrial septum:
- ostium secundum atrial septal defects (ASDs)
- patent foramen ovale (PFO) to reduce the risk of recurrent ischemic stroke in patients, predominantly between the ages of 18 and 60 years, who have had a cryptogenic stroke due to a presumed paradoxical embolism, as determined by a neurologist and cardiologist following an evaluation to exclude known causes of ischemic stroke.
DESCRIPTION:
- Implant (occluder) and delivery catheter (a small tube)
- Occluder made of self-expanding, nickel-titanium (Nitinol) wires, covered in woven fabric
- Occluder shaped as two discs connected in the cente
- Occluder compressed to a small size to allow it to pass through the delivery catheter for implantation
EFFECTIVENESS:
- Prospective, randomized (2:1), open-label, multi-center study, n=664, antiplatelet medical management and PFO closure with the GORE® CARDIOFORM Septal Occluder vs. antiplatelet medical management alone
- Co-primary endpoints: Freedom from recurrent stroke and incidence of new brain infarction. PFO closure was associated with a statistically significant
77% relative risk reduction in recurrent stroke - PFO closure was also associated with a statistically significant 49% relative risk reduction in incidence of new brain infarction
SAFETY:
- No significant difference in overall rate of SAEs between the control (medical management) and device groups
- Low rate of device- or procedure-related SAEs (3.6%)
- Subjects had a higher incidence of atrial fibrillation or flutter (6.6%), but were non-serious
- No device- or procedure related deaths.
REGULATORY PATHWAY: Supplemental PMA
- For expanding the indications to include closure of the patent foramen
ovale (PFO) to reduce the risk of recurrent ischemic stroke - Product Code: MLV
- Postapproval studies: Safety through 5 years post-procedure, acute, subacute, and longterm safety and effectiveness
REIMBURSEMENT:
- Approved CMS IDE study
- Partnership with CODING STRATEGIES (an independent industry-leading resource in coding, coverage, and reimbursement education)

GUARDIAN Connect System
Medtronic MiniMed, Inc.
INDICATION FOR USE: For continuous or periodic monitoring of glucose levels in the interstitial fluid under the skin, in patients (14 to 75 years of age) with diabetes mellitus.
Provides real-time glucose values and trends through a Guardian Connect app installed on a compatible consumer electronic mobile device. It allows users to detect trends and track patterns in glucose concentrations. The Guardian Connect app alerts if a Guardian Sensor (3) glucose level reaches, falls below, rises above, or is predicted to surpass set values.
The Guardian Sensor (3) glucose values are not intended to be used directly for making
therapy adjustments, but rather to provide an indication of when a finger stick may be
required. All therapy adjustments should be based on measurements obtained using a
home glucose monitor and not on values provided by the Guardian Sensor (3).
DESCRIPTION:
- Provides real-time glucose values and trends through a Guardian Connect app installed on a compatible mobile device platform (e.g., iPhone or iPad)
- Guardian Connect app is a mobile medical application that allows users to track patterns in glucose concentrations and to possibly identify episodes of low and high glucose
- System is designed to provide continuous glucose monitoring for up to seven days
- System consists primarily of a sensor, transmitter, and mobile medical app
EFFECTIVENESS, HUMAN FACTORS USABIITY, RISKS:
- Evaluate the performance of the Guardian Sensor (3) to support 7 days of use
- Missed alerts and false negative hypoglycemia and hyperglycemic readings related to patients not being alerted to the need to perform a fingerstick to detect hypoglycemia or hyperglycemia
REGULATORY PATHWAY: PMA
- Device Procode: MDS
- The Guardian Sensor (3) used with the Guardian Connect system is the same as the
Guardian Sensor (3) used with the MiniMed 670G System (P160017) and the iniMed
630G System (P150001/S008), which were previously approved
REIMBURSEMENT:
- Continuous Glucose Monitors are reimbursed by CMS
FDA News: Predictive Toxicology Roadmap, 2019 FDA Budget, Medical Device Safety Action Plan, Opioid Use Disorder Treatments, Drug Safety Priorities

Predictive Toxicology Roadmap
Significant steps by FDA to upgrade toxicology toolboxes
- Expand toxicology predictive capabilities
- Potentially reduce the use of animal testing
Goals of roadmap for integrating predictive toxicology methods into safety and risk assessments
- Develop and evaluate emerging toxicological methods and new technologies
- Incorporate these methods and technologies into regulatory review

Remarks from FDA Commissioner Scott Gottlieb, M.D. on Fiscal Year 2019 budget request for FDA
President’s 2019 Budget request for $5.8 billion in total resources for FDA
- Includes $190 million in user fees
- Requests new FDA resources to advance science, domestic technology, public health
- Advance new paradigm in regulation of digital health technology
- Advanced manufacturing to bring more production back to US
- Improve ability to respond to public health emergencies like flu
- Modernize generic drug review
- New approaches for treatments for rare pediatric diseases
Build knowledge management platform for drug and medical device review programs
- Store and manage collected experience of review staff
- Essential to modernizing medical product review and establish scientific precedents
- Issue guidances documents focused on specific diseases using efficient approaches
 Medical Device Safety Action Plan: Protecting Patients, Promoting Public Health
Medical Device Safety Action Plan: Protecting Patients, Promoting Public Health
Outlines how Agency will encourage innovation to improve safety, detect safety risks earlier, and keep doctors and patients better informed
- Robust medical device patient safety net
- Regulatory options to streamline and modernize timely implementation of postmarket mitigations
- Innovation towards safer medical devices
- Advance medical device cybersecurity
- Integrate premarket and postmarket offices and use of a Total Product Life Cycle (TPLC) approach to device safety
 New steps to encourage more widespread innovation and development of new treatments for opioid use disorder
New steps to encourage more widespread innovation and development of new treatments for opioid use disorder
Encouraging more widespread innovation and development of medication for use in medication-assisted treatments (MAT)
- Three FDA-approved MAT drugs – methadone, buprenorphine and naltrexone
- Facilitate development of new MAT products, and new formulations of existing drugs
FDA issued draft guidance: Opioid Dependence: Developing Buprenorphine Depot Products for Treatment
- Drug development and trial design issues
- Possible ways for innovations in buprenorphine products
- Use of 505(b)(2) regulatory pathway for product development programs
- Develop validated measurement of “craving” or “urge to use” illicit opioids

2017 Drug Safety Priorities
CDER’s efforts to enhance drug safety for the American public
- Safety surveillance and oversight of marketed drug products 7,446 safety reviews
- Importance of real-world evidence to help advance drug safety science: New scientific computing and data storage technologies to gain valuable information from “real world evidence.”
- New tools and new approaches for fighting our Nation’s opioid crisis: To 1) decreasing exposure and preventing new addiction, 2) safely treating those with opioid addiction, 3) developing safe and effective novel alternative therapies to opioids, 4) improving enforcement of safety measures and assessing benefit-risk ratios.
- Safety oversight for generic drugs: Flag early safety concerns
- Efforts to reduce preventable harm from medications: Safe Use Initiative
- Compounded drugs – continuing regulatory and oversight efforts: Conducted 140 inspections, sent 55 warning letters, and issued 40 recalls related to compounding.
- Diverse strategies, tools, and services for communicating drug safety: Responded to 57,094 inquiries from the public
Image credit: FDA
Spectrum of Disease Conditions
Spectrum of Disease Conditions
- Addiction
- Analgesia/Anesthesiology/Anti-inflammatory
- Cardiovascular Disease
- Dermatology
- Endocrinology/Metabolism/Bone
- Gastroenterology
- Hematology/Coagulation
- Immunomodulators
- Infectious Disease (viral)
- Infectious Disease (non viral)
- Medical Imaging
- Neurology
- Oncology
- Ophthalmology
- Psychiatry
- Pulmonary
- Renal Disease
- Rheumatology
- Urologic
Market Authorizations: ACUVUE OASYS Light Adaptive Contact Lens, IDx-DR retinal diagnostic software, OCS Lung System
 Acuvue Oasys Contact Lenses with Transitions Light Intelligent Technology
Acuvue Oasys Contact Lenses with Transitions Light Intelligent Technology
Johnson & Johnson Vision care
USE: Soft Contact lenses that automatically darkens the lens when exposed to bright light. Indicated for daily use to correct the vision of people with non-diseased eyes who are nearsighted (myopia) or farsighted (hyperopia).
ADDRESSING UNMET NEED: First contact lens to incorporate the same technology that is used in eyeglasses that automatically darken in the sun
DESCRIPTION:
- Contains photochromic additive that adapts the amount of visible light filtered to the eye based on the amount of UV light to which they are exposed
- Results in slightly darkened lenses in bright sunlight that automatically return to a regular tint when exposed to normal or dark lighting conditions.
SAFETY AND EFFECTIVENESS:
- Clinical study of 24 patients that evaluated daytime and nighttime driving performance while wearing the contact lenses
- No evidence of concerns with either driving performance or vision while wearing the lenses
- May cause inflammation or infection in or around the eye or eyelids
REGULTORY PATHWAY: 510(k)
- Classification: II
- Regulation No. : 886.5925
- Classification Product Code: LPL
- Subsequent Product Code: MVN
REIMBURSEMENT
- Medicare provides limited coverage for contact lenses under Medicare Vision Services
- Acuvue Brand covered by private payors
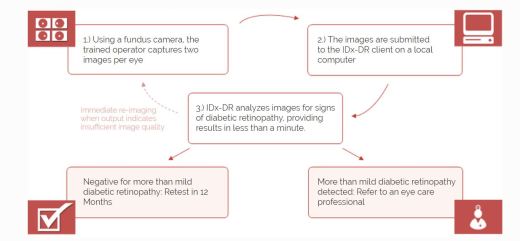
IDx-DR Retinal diagnostic software device
IDx LLC
INDICATION FOR USE: For use by health care providers to automatically detect more than mild diabetic retinopathy (mtmDR) in adults diagnosed with diabetes who have not been previously diagnosed with diabetic retinopathy. IDx-DR is indicated for use with the Topcon NW400.
ADDRESSING UNMET NEED:
- Diabetic retinopathy is the most common cause of vision loss among the more than 30 million Americans living with diabetes
- Leading cause of vision impairment and blindness among working-age adults
- First medical device to use Artificial Intelligence (AI) to detect greater than a mild level of the eye disease diabetic retinopathy in adults who have diabetes
DESCRIPTION:
- Software program using AI to analyze eye images taken with retinal camera, Topcon NW400
- Doctor uploads digital images of retinas to cloud server with IDx-DR software
- Software provides doctor with one of two results
- “more than mild diabetic retinopathy detected: refer to an eye care professional”
- “negative for more than mild diabetic retinopathy; rescreen in 12 months.”
- If positive result – further diagnostic evaluation and possible treatment as soon as possible
GENERIC DEVICE TYPE: Retinal diagnostic software device.
Prescription software device that incorporates an adaptive algorithm to evaluate ophthalmic images for diagnostic screening to identify retinal diseases or conditions.
ACCURACY & PRECISION:
- Clinical study of retinal images, n=900 patients with diabetes, 10 primary care sites
- Correctly identify presence of more than mild diabetic retinopathy: 87.4%
- Correctly identify patients who did not have more than mild diabetic retinopathy: 89.5%
IDENTIFIED RISK & MITIGATION MEASURE:
- False positive results leading to additional unnecessary medical procedures (Diagnostic algorithm failure, Software failure): Clinical performance testing;
Software verification, validation, and hazard analysis; Protocol for technical specification changes - False negative results leading to delay of further evaluation or treatment
(Diagnostic algorithm failure, Software failure): Clinical performance testing
Software verification, validation, and hazard analysis; Protocol for technical specification changes; Labeling - Operator failure to provide images that meet input quality specifications: Labeling,
Training, Human factors validation testing
REGULATORY PATHWAY: De Novo request
- Regulation No.: 21 CFR 886.1100
- Regulation Name: Retinal diagnostic software device
- Regulatory Class: Class II
- Product Code: PIB
REIMBURSEMENT:
- AI diagnostic approach could support CMS’ value-based reimbursement

Organ Care System (OCS) Lung System
TransMedics, Inc.
INDICATION FOR USE: Portable organ perfusion, ventilation, and monitoring medical device indicated for the preservation of standard criteria donor lungs in a near physiologic, ventilated, and perfused state for double lung transplantation.
DESCRIPTION:
- Lung Console: Non-sterile, reusable, portable enclosure housing electronic display and non-sterile mechanical/electrical elements to warm, pump, ventilate, and manage gas content of perfusate
- Lung Perfusion Set (LPS): Sterile, single-use perfusion module, organ chamber and circulatory system to perfuse and ventilate lung, facilitate management of fluids
- OCS™ Lung Solution: High oncotic solution used for ex-vivo flush and perfusion of donor lungs when combined with packed red blood cells (pRBCs)
EFFECTIVENESS AND SAFETY:
- Randomized, controlled, multi-center, international, prospective
clinical trial, OCS™ Lung System vs. current cold storage standard of care (SOC), n=407 - Primary Graft Dysfunction (PGD) grading, including reduced survival and
increased incidence of Bronchiolitis Obliterans Syndrome (BOS) - Patient survival at day 30 post-transplantation and ISHLT PGD3 within 72 hours post-transplantation
- No-inferiority vs. SOC, longer-term (2-year) survival and BOS rates comparable
- Similar lung graft-related serious adverse events (LGRSAEs) through 30 days post-transplantation
REGULATORY PATHWAY: PMA
- Class III, Product Code: QBA
- Priority Review
- Gastroenterology-Urology Devices Panel Meeting: Voted 11-2 that there is reasonable assurance the device is safe, 8-5 that there is reasonable assurance that the device is effective, and 9-4 that the benefits of the device do outweigh the risks
- Post-approval studies : Long-term patient outcomes, OCS Lung Thoracic Organ Perfusion (TOP) PAS Registry
Image credit: J&J, IDX, TransMedic
Model Informed Drug Development
 Model-Informed Drug Development Pilot Program
Model-Informed Drug Development Pilot Program
Pilot Program to facilitate the development and application of exposure-based, biological, and statistical models derived from preclinical and clinical data sources
- Quantitative methods to balance risks/benefits of drugs in development
- Can improve clinical trial efficiency, increase probability of regulatory success, optimize drug dosing/therapeutic individualization without dedicated trials
Goals of the MIDD Pilot Program
- Discuss application of MIDD approaches to development and regulatory evaluation of medical products
- Provide advice about how particular MIDD approaches can be used in a specific drug development program
FDA News: Medical device needs for rare diseases, Accelerating next generation sequencing-based tests, Essure sale retsriction
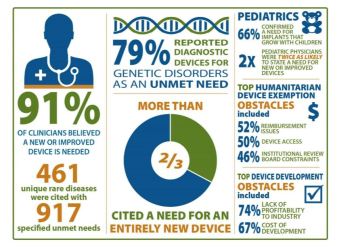
Unmet medical device Needs for patients with rare diseases
FDA, National Center for Advancing Translational Sciences (NCATS)/Office of Rare Diseases Research (ORDR) at NIH sought to better understand medical device needs of patients with rare disease
- Generate meaningful data to inform patients, practitioners, policymakers, and device developers
- Needs, barriers, and incentives
Online survey of four clinician groups
- satisfaction with current devices
- unmet needs for specific rare diseases
- impediments to medical device development
Survey Respondents: 1,342 clinicians
Findings
- Patients with rare diseases face numerous unmet needs
- Device needs of pediatric patients – grow with child, be modified to smaller size, less invasive
- Creating entirely new devices needed vs. modifying/repurposing existing devices
- Limitations included lack of sensitivity/specificity, cumbersome and invasive
- Costs of research, lack of profitability, challenges of conducting trials are challenges

FDA finalizes guidances to accelerate the development of reliable, beneficial next generation sequencing-based tests
Finalized two guidances for efficient development of novel technology that scans DNA to diagnose genetic diseases-next generation sequencing (NGS)
- Recommendations for designing, developing, and validating tests; for continued advancement of individualized, genetic-based medicine
- Modern and flexible framework
- Reliance on clinical evidence from FDA-recognized public databases to support clinical claims e.g. ClinGen
- Recommendations for designing, developing, validating tests to diagnose individuals with suspected genetic diseases
Based on extensive feedback from the public and stakeholders; continuation of creating regulatory efficiencies in the development and review of NGS tests
 FDA Restricts the Sale and Distribution of Essure
FDA Restricts the Sale and Distribution of Essure
Order to restrict the sale and distribution of the Essure device
- Ensure all women provided with adequate risk information so that they can make informed decisions
- Taking this step because some women were not being adequately informed of Essure’s risks before getting the device implanted
- Boxed Warning including perforation of the uterus and/or fallopian tubes, identification of inserts in the abdominal or pelvic cavity, persistent pain, and suspected allergic or hypersensitivity reactions
- FDA closely evaluating new information on the use of Essure; requires additional, meaningful safeguards to ensure women are able to make informed decisions
New Essure labeling
- Restricts sale and distribution to only health care providers and facilities that provide information to patients about the risks and benefits of this device
- Review “Patient-Doctor Discussion Checklist – Acceptance of Risk and Informed Decision Acknowledgement”
- Patient and physician required to sign
- FDA will review and monitor Bayer’s plan to ensure compliance of restriction
Image credit: FDA
Help FDA improve MedWatch System
 Help FDA Improve FDA MedWatch Forms
Help FDA Improve FDA MedWatch Forms
MedWatch is FDA gateway for clinically important safety information and reporting serious problems with human medical products
- Physicians and Consumers can report unexpected side effects, adverse events, or other problems through MedWatch program
- Report adverse events, product problems, errors with use, evidence of therapeutic failure is suspected or identified
FDA is seeking comments to help improve adverse event information collection
- Forms 3500, 3500A and 3500B
Image credit: FDA
CDRH Device Evaluation Intern Program

Device Evaluation Intern Program
Challenging and rewarding experience for individuals interested in pursuing careers in the fields of science, engineering, and/or medicine
- Test educational interests in practical work environment
- Gain professional “real work” experience
- Work alongside Agency’s top healthcare authorities, establish professional contacts
Image credit: FDA
Benefit-Risk Assessment in Drug Regulatory Decision-Making : 2018-2022

Benefit-Risk Assessment in Drug Regulatory Decision-Making
Under FDARA 2017, FDA committed to furthering implementation of structured benefit-risk assessment into drug review
Commitments on Enhancing Benefit-Risk Assessment
- Continue implementation of the Benefit-Risk Framework
- Participate in stakeholder meetings
- Draft guidance on benefit-risk assessment
- Revise relevant MAPPs and SOPPs
- Conduct second evaluation of Benefit-Risk Framework
Additional Opportunities to Enhance FDA’s Benefit-Risk Assessment
- Improving accessibility for approved products
- Use to support Advisory Committee meetings
- Exploring additional tools to support assessment
Image credit: FDA